

近年来,科研工作者一直在寻找能够快速、简捷、高效发现药物、催化剂、蛋白质等新材料和新反应途径的自动化合成系统.人工智能(Artificial Intelligence, AI)和机器学习(Machine Learning, ML)的出现使实现这一目标成为可能.ML是AI和计算机科学的一个重要分支,它研究和构建的是一种特殊算法而非某一个特定的算法,能够让计算机自身从数据中学习并进行下一步预测[1].ML算法能够从大量化学数据中寻找规律和联系,帮助科研工作者做出更合理的判断和决策, 加快研究过程.ML在化学合成领域[2-3]的应用已经取得许多令人瞩目的成果,如分析化学反应进行反应优化[4⇓-6]、逆合成分析寻找产物的最佳合成路径[7-8]、比较药物活性辅助药物设计[9]等.ML正在成为除分子模拟之外的计算化学的新范式.ML为化学合成领域的发展带来无限生机的同时,也为合成研究带来了新的难题与挑战.
1 ML应用于化学合成的基本流程
图1

图1
在化学科学中应用ML算法的工作流程
Fig.1
Workflows for applying ML algorithms in chemical sciences
1.2 特征化数据
输入数据的形式往往影响ML的效果,常见的输入数据形式主要有向量、矩阵和图像3种.由于化学数据的特殊性,绝大多数数据无法直接作为模型的输入,需要进行数据转换.将原始数据转换成更适合算法处理的格式,这一过程称为特征化或特征工程.在化学领域中,使用分子描述符对分子信息进行描述表示.分子描述符是指分子在某一方面性质的度量,既可以是分子的物理化学性质,也可以是根据不同算法推导出来的与分子结构相关的数值指标.选择出与研究对象最密切相关的描述符对整个ML过程而言十分重要,表1总结了常见的分子描述符.
表1 常见的分子描述符总结(以苯酚乙酯为例)
Tab.1
| 描述符名称 | 表现形式 | 优势 | 不足 | 适用范围 |
|---|---|---|---|---|
| SMILES[13]字符 SMARTS[14]字符 Inchl字符 | SMILES: CC(OC1=CC=CC=C1)=O SMATRS: [C]-[C](-[O]-[C]1: [C]: [C]: [C]: [C]: [C]: 1)=[O] Inchl: 1S/C8H8O2/c1-7(9)10-8-5-3-2-4-6-8/h2-6H, 1H3 | 采用线性方法对分子进行表示,简单易操作;不同分子的SMILES不同,具有唯一性;占用内存小,节省存储空间. | 丢失分子的三维信息;每个SMILES字符串对分子图的表示方法不唯一,即可从不同方向对分子图进行编码. | 不需要分子空间信息;需要大量数据进行训练的模型. |
| 分子指纹 | 图示① | 采用比特量形式表示分子,编解码简单;能够表示分子的局部信息;分子的特征之间相互独立. | 分子信息存在冗余,占用存储空间大;计算时间长,每次计算需要进行遍历. | 擅长计算分子之间的相似性;描述分子的部分结构信息. |
| 分子图 | 图示② | 分子可视性强;描述符可解释性强;能够描述分子的三维信息. | 信息传递更新过程慢,计算过程复杂. | 图神经网络模型的输入;需要分子空间信息的场合. |
| 量子化学描述符 | 过渡态能量[15]、波函数、Fukui函数[16]、分子键序、分子电荷等. | 能够精准计算分子的化学和物理性质. | 计算时间长;计算过程繁琐. | 需精确描述分子性质的场合. |

其中,需要特别注意量子化学描述符,这类描述符一般通过Gaussian、NWChem等软件计算获得,能够较准确地描述分子的化学和物理性质,计算结果具有较高的可靠性.此类描述符不仅是ML的输入数据形式之一,而且能够作为化学实验结果的佐证工具,如张书宇等[15]总结了密度泛函理论(Density Functional Theory, DFT)计算验证轴向手性苯乙烯合成的机理和方法.首先,他们发现镍催化对映选择性三分量自由基传递烯烃还原偶联可以实现轴向手性苯乙烯的合成,可以借助DFT计算寻找反应中的过渡态对整个反应机理进行佐证[17].其次,使用DFT计算非常规远程杂芳基迁移对非活性烯烃进行异芳基氟烷基化过程中杂芳基迁移后氢原子转移(Hydrogen Atom Transfer, HAT)的溶剂化自由能,计算结果与实验数据吻合,与反应机理相印证[18].
1.3 模型训练
数据特征和算法性能决定了ML的有效性和正确率,不同的ML算法产生的结果不同,甚至同一种ML算法结果也会因数据特征而异.算法是ML过程的关键,选择时需从实际问题出发,多方面综合考虑.目前,根据算法特征将ML分为4类[19]:监督学习(Supervised Learning, SL)、无监督学习(Unsupervised Learning, UL)、半监督学习(Semi-Supervised Learning, SSL)和强化学习(Reinforcement Learning, RL).
1.4 分析结果
待模型训练完,可以将真实实验数据与预测数据进行对比来评估模型质量,分析模型学习结果能否较好解决实际问题.针对不同问题,需采用不同的模型评估指标,如评估SL中分类模型可以采用准确率(Accuracy)、召回率(Recall)、受试者工作特征曲线 (Receiver Operating Characteristic, ROC)等;评估SL中回归模型时可以借助平均绝对误差(Mean Absolute Error, MAE)、均方误差(Mean Square Error, MSE)、均方根误差(Root Mean Square Error, RMSE)、决定系数(R2)等.
2 ML在化学合成及表征领域的应用
2.1 随机森林的应用
随机森林(Random Forest, RF)是一种集成分类算法,由 Breiman[34]提出,使用“并行”决策树(Decision Tree, DT)的方式,如图2所示.DT模型是一种以树结构为依据的分类算法,由节点和分支组成.从树的根节点开始,依次向下分类.一棵DT有且仅有一个根节点.能够将一个复杂的决策过程分解成一组更简单的决策,从而提供一个通俗易懂、易解释的解决方案是DT模型最大的优势[35].在RF中,每棵DT生成一个随机向量,向量之间相互独立且分布相同,根据一定的投票机制或取平均值得到最佳分类结果.RF由多棵DT组合生成的,因此该算法能够最大限度地减少过拟合问题,提高预测精度和控制力[36].
图2
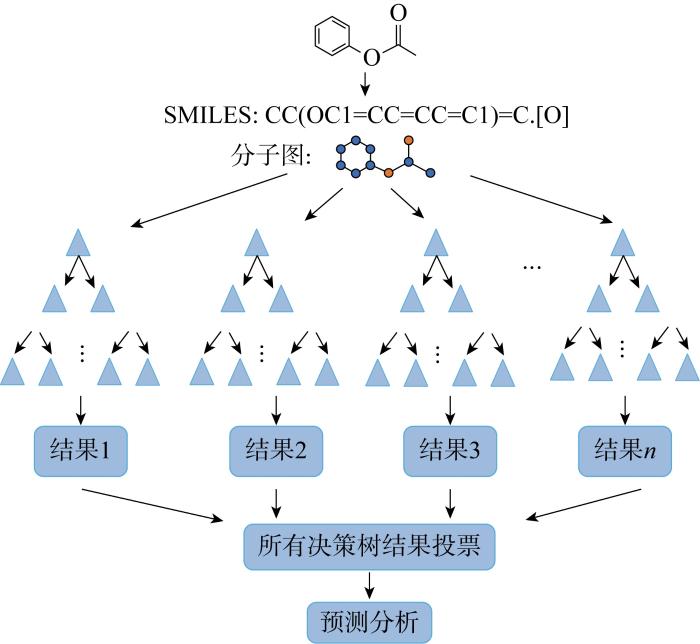
图2
输入数据形式以分子图和SMILES字符为例的RF模型
Fig.2
RF of molecular graph and SMILES as input data forms
RF作为一种集成算法,具有良好的鲁棒性和可靠性,分类效果好,适合作为基线模型进行产率预测.高通量实验结合DFT计算能够缩短RF模型建立的时间,使得该模型在化学合成领域得到广泛应用.RF解决的主要是分类问题,分类问题是数据挖掘处理的一个重要组成部分,目标是根据已知样本的某些特征,判断新样本属于哪种已知的样本类.科研工作者通常从预测精确度、计算复杂度、模型简洁度对多种分类算法进行比较评价.Singh等[35]选择了5种不同的轴向手性联萘催化剂共368个不对称氢化反应和一系列烯烃、亚胺作为训练数据集生成了一个RF模型,如图3(a)所示,选择能够共享等效或具有共同核心区域的反应参数作为输入,以对映体过量百分率(ee%)作为输出值,对输入化合物进行分类,每种催化剂生成一棵DT,形成RF模型.与其他模型相比,RF模型得到了较高的精度,表明RF模型在识别不对称催化反应时有良好的应用.Kang等[37]设计了一种RF模型用于预测分子的激发能量和相关振荡器强度,首先使用RDkit工具包计算出分子的扩展连通性指纹(Extended-Connectivity Fingerprints,ECFP)、MACC键等分子描述符作为输入数据生成许多DT模型,对所有DT的预测结果进行投票选择评估,生成RF模型.该模型使用了近50万个DFT数据进行训练,实验结果表明RF模型预测振荡器强度和有机化合物最高强度跃迁激发能的精准度最佳.Li等[38]报道了一种物理有机特征描述符和RF相结合的模型(PhyOrg-RF),对杂环自由基C—H官能团的区域选择性进行预测.在样本外测试集中PhyOrg-RF模型实现了94.2%的位点预测精度和89.9%的选择性预测精度,拥有较好的区域选择性预测能力,使用其他已公开实验数据进行测试验证了PhyOrg-RF具有优异的泛化能力.Ahneman等[39]提出一种基于RF的预测钯催化Buchwald-Hartwig胺化反应产率的模型,如图3(b)所示.其中,10 mol%表示催化剂与反应物的物质的量之比为10%.通过高通量实验生成 4608 个反应数据,将简单原子、分子和振动描述符作为训练集进行模型训练.该模型的测试集RMSE为7.8%,R2为0.92,该模型未曾出现过拟合现象,能够以RMSE为11.3%、R2=0.83的精度成功预测反应产率.Tomberg等[16]选择RF作为分类模型,判断芳香类化合物的反应位点,与人工神经网络(Artificial Neural Network, ANN) 、LR和SVM模型相比,RF不仅训练时间短,而且正确率高达93%.Xu等[40]提出一种将过渡态知识模型与额外树(Extra Tree, ET)模型相结合的方式,对钯电催化C-H活化的对映选择性预测.RF在一个随机子集内得到最佳分类属性,而ET完全随机得到分类属性,同时具有随机性和最优性.
图3
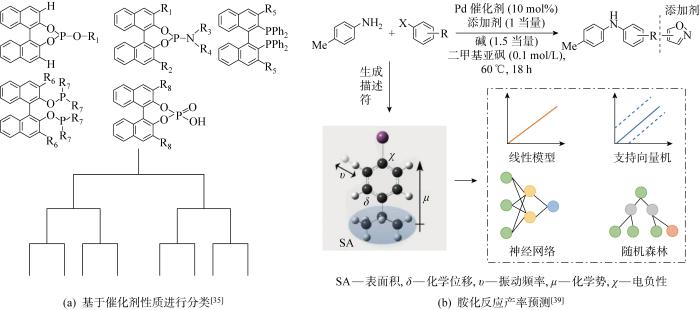
图3
催化剂分类形成RF模型过程和RF作为比较模型的反应产率预测
Fig.3
Process of catalyst classification to form RF model and prediction of reaction yield using RF as a comparative model
2.2 卷积神经网络的应用
式中:
CNN主要用于解决SL中的回归问题.回归问题研究的是自变量和多个变量之间的关系,用于处理离散型数据.NN受人类大脑的启发,模仿生物神经元信号相互传递的方式,能够无限逼近非线性模型,在化学合成领域有着出色的表现.首先,CNN借助共享卷积核的方式降低计算复杂度,可以快速处理高维化学数据.Hirohara等[47]设计一种SMILES字符串与CNN相结合的模型(SCFP),用于化学基序检测.使用TOX21数据集中分子的SMILES字符串,将字符串输入CNN中,其中卷积操作只在SMILES字符串的一个方向进行,如图4(b)所示(k1和k2表示滤波器),由受试者工作特征及其曲线面积(ROC-AUC)进行评分.此模型还可以被视作一种分子指纹,在SR-MMP子数据集的化学空间中比ECFP分子指纹表达效果好.选取NR-AR子数据集进行化学基序分析成功检测出一种类固醇样化学基序.Wallach等[48]报道了一种基于CNN的模型AtomNet,预测药物发现应用中小分子的生物活性.AtomNet有两点优势:①CNN的强制局部性与化学基团之间相互作用时产生的局部效应相吻合;②将有关配体的信息和相关目标结构的信息相结合,十分适合结构的亲和力预测,并且选择原子在靶位结合点的位置,能够让模型发现任意分子特征.选择文档理解数据集和评估(DUDE)基准的数据集,评估数据集的AUC及其对数值,AtomNet中 57.8%目标的AUC大于0.9.Hughes等[49] 使用702个环氧化反应数据训练了一种CNN模型,在环氧化位点识别上表现出0.949 的AUC结果,在区分环氧化分子上表现出0.793 的AUC结果.此网络不仅能够预测分子的环氧化作用,还能预测分子中的环氧化作用位点.该课题组还将类似的模型应用到了小分子与软亲核试剂的反应预测中,对是否能够发生反应进行预测,准确率为80.6%,小分子反应位点的预测准确率达到了90.8%[50].
图4
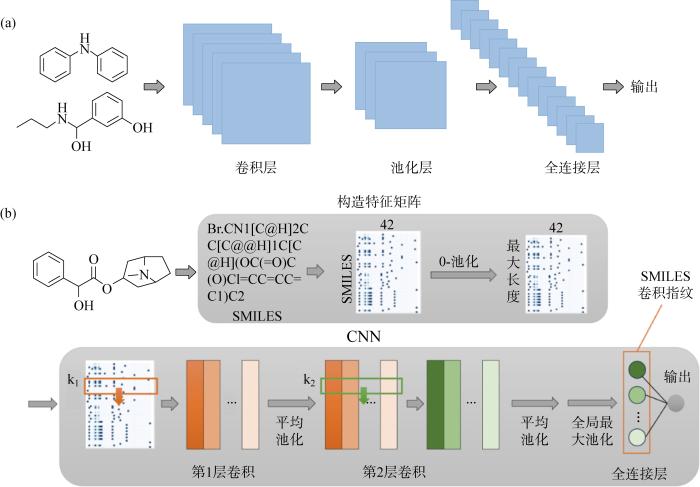
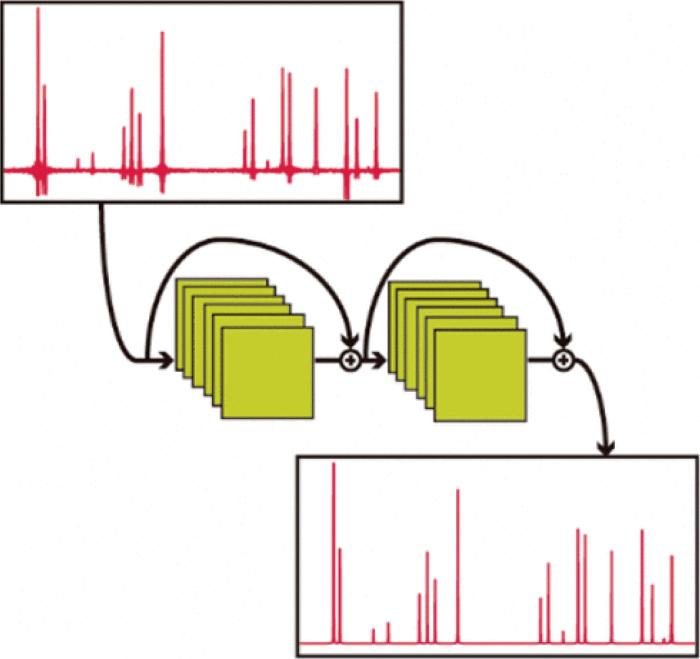
CNN不仅可以对高维数据进行快速降维,在图像处理如图谱分析方面也有不可比拟的优势.Xing等[51]提出一种基于CNN的生物学驱动代谢组学习工作流程SteroidXtract,可实现在非靶向代谢组学数据集中对类固醇化合物二级质谱谱图(MS2)的自动化快速索取.SteroidXtract是一种高灵敏度、高特异性提取类固醇化合物谱图的工具,该方法不使用传统统计驱动的代谢组学习数据处理过程,更加高效简洁.Zheng等[52]借助CNN得到一种快速获取高质量核磁共振纯位移谱的新方法,如图5所示.通过在实验中引入指数采样来加速PSYCHE纯位移谱的获取,使用CNN对欠采样的图谱进行重建,可以在低采样率的情况下获得干净的纯位移谱.卷积核的选择对CNN算法的成败有着关键作用,通常选择大小为3×3,步长为1的卷积核.此外,也可根据实际应用进行调整,但需注意的是,卷积核尺寸越大、步长越大,得到的特征图数量越少,提取出的特征数目越少,可能会影响后续预测的准确性.共享卷积核使得CNN算法复杂度大大降低,因此当处理大量高维数据时,推荐使用CNN算法.
图5
2.3 图神经网络的应用
几何深度学习[53]的出现将NN模型扩展到了非欧氏空间.图神经网络(Graph Neural Network, GNN)是处理非欧氏空间数据的常用模型,能够以递归形式合并邻近节点的信息或消息,同时自然地捕获图形结构和节点特征[54].GNN通过图节点之间的消息传递捕获图中重要信息, 查看相邻节点上的信息来确定每个节点的最终状态,以迭代方式传播相邻节点信息来学习目标节点的特征,直至到达稳定的固定点.简单来讲,GNN获取信息的过程可以概括为:聚合—更新—循环,如图6所示.首先使用某种方法对节点信息f1~f5进行表征描述,使每一个节点学习一个嵌入状态,这个状态用来产生所需要的输出即更新后的节点信息f1,new~f5,new.给定节点和边的特征即可不断更新节点状态并获得最终输出.当所有节点的状态都趋于稳定状态时,节点的状态向量都包含了其邻居节点和相连边的信息,需要保证整个更新过程收敛.
图6
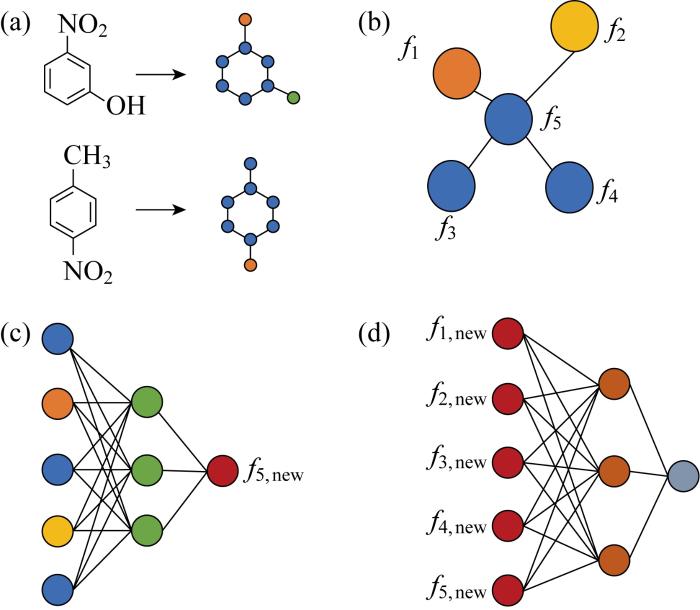
2.4 UL和RL的应用
RL在逆合成设计中采取不确定性下的决策,不仅比传统方法处理速度快,而且置信度更高.RL通常以马尔可夫决策过程为框架[65],奖励函数为核心,奖励函数决定了主体通过动作学习实现的目标.奖励函数可以是离散的也可以是连续的.Segler等[66]设计了一种将蒙特卡洛树和NN相结合的逆合成分析方法,由计算机辅助合成设计程序(Computer-Aided Synthetic Planning, CASP)生成合成路线,使用蒙特卡洛树和3个不同的NN进行搜索.从目标分子开始,选择树中最有可能的下一个位置,直至到达叶节点.通过扩展策略预测可能出现的叶节点的子节点,并将其添加在树中,对推出过程进行评估.结果的位置值表示RL更新其树搜索策略所需要的奖励,找到解决方案会收到奖励,找到部分解决方案会收到部分奖励,未找到方案则会收到惩罚.不断迭代更新直到达到最大的时间或迭代次数,通过选择具有最高位置值的断开路径来决定最终的合成路线,具体搜索过程如图7所示,包括选择最可能的位置、使用扩展程序对节点进行扩展、选择评估新节点和更新4个阶段,其中T1~Tn为所有可能的概率分布,R1~Rk表示完整的反应物.
图7

3 挑战与机遇
3.1 建立高质量数据集和模型评估标准
数据集质量直接决定了ML模型训练的成败,构建大型数据集是一个耗时且费力的过程,因此这些数据集的共享访问对整个化学界都很重要.寻找化学数据之间客观联系的前提是拥有足够量的数据,但化学数据并非像图像数据一样简单易得且具有良好的通用性.数据量不足很容易导致ML训练失败,无法产生所需结果.在化学合成领域中底物和催化剂的微小改变都会导致合成产物的不同,因此有效数据少之又少.当前开源化学数据集涵盖的化学类型不多、配体种类并不全面,如广泛应用于图像处理领域的ImageNet数据库[67]和涵盖诸多量子化学、物理化学信息数据的MoleculeNet数据库[68],以及收集了大量小分子化合物量化信息的GDB-17[69]及其子库QM8、QM9等,在模型训练方面均具有显著的成效.获得大量高质量数据,建立完备数据集才有可能最大限度地发挥ML在化学领域的潜力.特别是DeepChem、SchNetPack[70]等软件的发展,解决高质量数据的问题与计算化学软件包的发展息息相关.
3.2 增强ML结果与实验数据的匹配
长期以来,ML模型复杂度已从线性上升到ANN.ML在化学合成领域应用更广泛是因为可以借助高通量实验或模拟研究等方式获得大量数据,辅助化学家进行产物、产率的预测,减少人力、物力的投入.然而,ML在化学合成中的应用仍然有限.虽然当前NN算法可以无限逼近非线性模型,但需要大量训练数据作为支撑,并且它能处理的数据空间有限,无法在广阔的化学空间中做到处处预测精准.一种模型有时只能针对一种特定的化学反应,在一些实际应用中并不能寻找到最佳决策.因此,增强ML模型在化学合成领域的通用性是当务之急.ML自身的可解释性不强导致研究者需要基于化学知识对模型输出结果进行解释,但有时仍会出现不具有物理意义的结果.增强模型的可解释性既可以帮助研究者更好理解模型的输出和实际意义,也能帮助研究者更快掌握模型相关信息.
在化学合成领域中,ML特别是SL一直使用黑盒方法,但黑盒方法在可解释性、通用性、可靠性方面存在缺陷.这些缺陷很有可能会限制ML的应用,有时甚至产生错误的预测结果.ML与化学实验相结合有望生成具有更好可解释性、更高预测精度、更强通用性的模型.经过化学实验验证能够及时修改训练模型中的参数设置,以期达到最佳预测结果.
3.3 描述符和ML算法的发展
欲使ML方法预测的准确性得到进一步提高,分子描述符转换时要尽可能减少有效特征损失.描述符对ML的重要性不言而喻,目前建立描述符的方式共有4种:① 使用已有的SMILES字符串、分子指纹、分子图等;② 借助Python工具包生成描述符,RDkit是常用的工具包,包含分子指纹及其相关性的计算、分子三维表示等模块;③ 使用Gaussian等量子化学软件进行DFT计算,计算分子物理化学性质,将物理化学性质进行组合生成描述符;④ 根据反应特点,自行建立描述符. 现在已有的描述符生成方式均基于化学知识生成,如Zahrt等[64]提出一种平均空间占有率的描述符,分析不同催化剂在空间中的分布,有利于后续催化剂筛选.有效的描述符能够在数据集较小的情况下获得相对较好的预测结果.未来,研究者可以改进用于获得描述符的计算方法,采取半经验方法快捷、高效地生成高质量描述符;或许还可以将化学知识与ML相结合以及将基于化学知识的模型和数据驱动模型相结合生成描述符.
在ML领域,一个基本的定理为“没有免费的午餐”.换言之,没有一种算法可以完美地解决所有问题,尤其是对于SL的算法而言,如NN算法不是在任何情况下都比RF算法有优势,反之亦然.数据集的形式或规模都会对算法产生影响,因此,科研工作者应当根据实际需求选择合适的算法,即选择正确的ML任务.不同ML算法的使用范围和应用示例如表2所示.未来,期望ML算法能够增强其通用性和可解释性.
表2 ML在化学合成及表征领域的应用
Tab.2
| ML算法 | 注意事项/适用范围 | 应用实例 |
|---|---|---|
| SL-回归 | 研究自变量和其他变量之间的关系.一般使用不同模型进行拟合和交叉验证获得最优模型,具有很强的鲁棒性和容错性.需要考虑变量之间的相关性时采用多层回归,常用的NN模型能够无限逼近复杂的非线性模型,并行处理能力强,但是需要大量数据,输出结果的可解释性较弱. | 逻辑回归:预测催化反应产率[73] 多元LR:预测不对称反应中的对映选择性的关键参数[74⇓-76] 多种回归模型进行比较:预测反应产率[77] NN:正向反应预测[78] NN:从反应底物预测产物[5] NN:预测有机分子亲核性[79] |
| SL-分类 | 对待测数据进行分类,通常是几种算法之间比较评估得出最优模型用于后续预测分析.RF算法可以保证分类节点特征的最优性但要避免过拟合现象,ET算法可以使节点特征选择具有随机性和最优性.SVM算法可以处理非线性数据,但需要进行线性化操作,将其转化为高维线性数据. | RF、SVM、ANN:预测有机分子的水溶性[80] RF、SVM:预测交叉偶联反应各种潜在抑制配体的反应性能[39] RF:有机化合物的紫外-可见光谱分类[81] RF:设计催化剂[82] RF:预测反应类型[83] ET、RF:不对称区域选择性预测[40] |
| 贝叶斯理论 | 以贝叶斯公式为核心,模型容易理解,对小规模数据表现良好,过程简单,适合于小规模数据集的多分类问题,需要注意使用该理论时要有独立分布的假设前提. | 贝叶斯优化器:优化反应条件[84] 贝叶斯图卷积网络:预测分子表皮生长因子受体抑制活性[85] 贝叶斯学习:预测金属位点反应性质[86] |
| RL | 从环境中学习信息,比其他方法更加智能化.奖励函数的设计是整个过程的核心,适合用于反应条件的优化问题,但采样数据的效率不高. | RL:迭代化学反应结果,优化化学反应[87] 分子图+RL:分子设计[88] SMILES字符串+RL:药物设计合成[89] 分子图+RL:药物设计合成[90] |
4 结语
ML强大的数据处理能力为人们提供了一条更好理解分子性质、结构的新途径,在化学领域中得到了广泛应用.在不久的将来, ML算法的快速发展无疑将扩大可用于解决典型化学任务数据处理方法的储备.目前在化学合成及表征领域,并不存在通用性好、可解释性强、精度高的模型.无论ML模型效果多么优异,它只能提供相关性,并没有因果关系.为解决上述问题,每个ML模型特别是需要借此得出结论的,均需要相关化学知识进行严格验证,确保模型没有出现过拟合等不良现象.如今,ML在化学合成及表征领域应用广泛,但如何增强模型通用性、建立模型评估标准、完备开源数据集、将ML与实验相结合以及寻找更好的描述符仍是ML在化学合成及表征领域未来发展的重大挑战.未来,ML在化学研究中的应用会持续增加,化学工作者有必要了解相关模型背后的理论框架,找到ML和化学知识之间的交叉融合点.相信在不久的未来,以ML为代表的AI技术的引入和贯通应用将对化学合成及表征领域的发展做出不可磨灭的贡献.
参考文献
Machine learning: Trends, perspectives, and prospects
[J].
DOI:10.1126/science.aaa8415
PMID:26185243
[本文引用: 2]

Machine learning addresses the question of how to build computers that improve automatically through experience. It is one of today's most rapidly growing technical fields, lying at the intersection of computer science and statistics, and at the core of artificial intelligence and data science. Recent progress in machine learning has been driven both by the development of new learning algorithms and theory and by the ongoing explosion in the availability of online data and low-cost computation. The adoption of data-intensive machine-learning methods can be found throughout science, technology and commerce, leading to more evidence-based decision-making across many walks of life, including health care, manufacturing, education, financial modeling, policing, and marketing. Copyright © 2015, American Association for the Advancement of Science.
Machine learning for molecular and materials science
[J].DOI:10.1038/s41586-018-0337-2 [本文引用: 1]
RetroPrime: A Diverse, plausible and Transformer-based method for Single-Step retrosynthesis predictions
[J].DOI:10.1016/j.cej.2021.129845 URL [本文引用: 1]
Molecular transformer: A model for uncertainty-calibrated chemical reaction prediction
[J].
DOI:10.1021/acscentsci.9b00576
PMID:31572784
[本文引用: 1]
Organic synthesis is one of the key stumbling blocks in medicinal chemistry. A necessary yet unsolved step in planning synthesis is solving the forward problem: Given reactants and reagents, predict the products. Similar to other work, we treat reaction prediction as a machine translation problem between simplified molecular-input line-entry system (SMILES) strings (a text-based representation) of reactants, reagents, and the products. We show that a multihead attention Molecular Transformer model outperforms all algorithms in the literature, achieving a top-1 accuracy above 90% on a common benchmark data set. Molecular Transformer makes predictions by inferring the correlations between the presence and absence of chemical motifs in the reactant, reagent, and product present in the data set. Our model requires no handcrafted rules and accurately predicts subtle chemical transformations. Crucially, our model can accurately estimate its own uncertainty, with an uncertainty score that is 89% accurate in terms of classifying whether a prediction is correct. Furthermore, we show that the model is able to handle inputs without a reactant-reagent split and including stereochemistry, which makes our method universally applicable.Copyright © 2019 American Chemical Society.
Neural networks for the prediction of organic chemistry reactions
[J].Reaction prediction remains one of the major challenges for organic chemistry and is a prerequisite for efficient synthetic planning. It is desirable to develop algorithms that, like humans, "learn" from being exposed to examples of the application of the rules of organic chemistry. We explore the use of neural networks for predicting reaction types, using a new reaction fingerprinting method. We combine this predictor with SMARTS transformations to build a system which, given a set of reagents and reactants, predicts the likely products. We test this method on problems from a popular organic chemistry textbook.
Linking the neural machine translation and the prediction of organic chemistry reactions
[EB/OL].(
Retrosynthetic reaction prediction using neural sequence-to-sequence models
[J].
DOI:10.1021/acscentsci.7b00303
PMID:29104927
[本文引用: 1]
We describe a fully data driven model that learns to perform a retrosynthetic reaction prediction task, which is treated as a sequence-to-sequence mapping problem. The end-to-end trained model has an encoder-decoder architecture that consists of two recurrent neural networks, which has previously shown great success in solving other sequence-to-sequence prediction tasks such as machine translation. The model is trained on 50,000 experimental reaction examples from the United States patent literature, which span 10 broad reaction types that are commonly used by medicinal chemists. We find that our model performs comparably with a rule-based expert system baseline model, and also overcomes certain limitations associated with rule-based expert systems and with any machine learning approach that contains a rule-based expert system component. Our model provides an important first step toward solving the challenging problem of computational retrosynthetic analysis.
Towards "AlphaChem": Chemical synthesis planning with tree search and deep neural network policies
[EB/OL]. (
Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC)
[EB/OL]. (
Mobile data science and intelligent apps: Concepts, AI-based modeling and research directions
[J].DOI:10.1007/s11036-020-01650-z [本文引用: 1]
Cybersecurity data science: An overview from machine learning perspective
[J].
DOI:10.1186/s40537-020-00318-5
[本文引用: 1]
In a computing context, cybersecurity is undergoing massive shifts in technology and its operations in recent days, and data science is driving the change. Extractingsecurity incident patternsor insights from cybersecurity data and building correspondingdata-driven model, is the key to make a security system automated and intelligent. To understand and analyze the actual phenomena with data, various scientific methods, machine learning techniques, processes, and systems are used, which is commonly known as data science. In this paper, we focus and briefly discuss oncybersecurity data science, where the data is being gathered from relevant cybersecurity sources, and the analytics complement thelatest data-driven patternsfor providing more effective security solutions. The concept of cybersecurity data science allows making the computing process more actionable and intelligent as compared to traditional ones in the domain of cybersecurity. We then discuss and summarize a number of associatedresearch issues and future directions. Furthermore, we provide amachine learningbasedmulti-layered frameworkfor the purpose of cybersecurity modeling. Overall, our goal is not only to discuss cybersecurity data science and relevant methods but also to focus the applicability towards data-driven intelligent decision making for protecting the systems from cyber-attacks.
A short review of chemical reaction database systems, computer-aided synthesis design, reaction prediction and synthetic feasibility
[J].
SMILES, a chemical language and information system. 1. introduction to methodology and encoding rules
[J].
AMBIT-SMARTS: Efficient searching of chemical structures and fragments
[J].
DOI:10.1002/minf.201100028
PMID:27467262
[本文引用: 1]
We present new developments in the AMBIT open source software package for efficient searching of chemical structures and structural fragments. AMBIT-SMARTS is a Java based software built on top of The Chemistry Development Kit. The AMBIT-SMARTS parser implements the entire SMARTS language specification with several syntax extensions that enable support for custom modifications introduced by third party software packages such as OpenEye, MOE and OpenBabel. The goal of yet another open-source SMARTS parser implementation is to achieve better performance and compatibility with multiple existing flavours of the SMARTS language, as well as to provide utilities for running efficient SMARTS queries in large structural databases. We describe a combination of approaches towards lowering the computational cost and improving the response time of substructure queries. An exhaustive comparison of the AMBIT algorithm with several subgraph isomorphism implementations is performed. To demonstrate the performance of the entire system from an end-user point of view, response time statistics for Web service substructure search queries against a database of 4.5 M structures are also reported. The package has wide applicability in the implementation of various chemoinformatics tasks. It has already been used in several projects dealing with descriptor calculation and predictive algorithms, database queries, web applications and web services. Copyright © 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Synthetic strategies and mechanistic studies of axially chiral styrenes
[J].DOI:10.1016/j.checat.2023.100594 URL [本文引用: 2]
A predictive tool for electrophilic aromatic substitutions using machine learning
[J].DOI:10.1021/acs.joc.8b02270 URL [本文引用: 2]
Ni-catalyzed, enantioselective three-component radical relayed reductive coupling of alkynes: Synthesis of axially chiral styrenes
[J].DOI:10.1016/j.checat.2022.09.020 URL [本文引用: 1]
Electroreductive fluoroalkylative heteroarylation of unactivated alkenes via an unconventional remote heteroaryl migration
[J].DOI:10.1016/j.xcrp.2023.101385 URL [本文引用: 1]
Machine learning: Algorithms, real-world applications and research directions
[J].DOI:10.1007/s42979-021-00592-x [本文引用: 1]
Practical selection of SVM parameters and noise estimation for SVM regression
[J].We investigate practical selection of hyper-parameters for support vector machines (SVM) regression (that is, epsilon-insensitive zone and regularization parameter C). The proposed methodology advocates analytic parameter selection directly from the training data, rather than re-sampling approaches commonly used in SVM applications. In particular, we describe a new analytical prescription for setting the value of insensitive zone epsilon, as a function of training sample size. Good generalization performance of the proposed parameter selection is demonstrated empirically using several low- and high-dimensional regression problems. Further, we point out the importance of Vapnik's epsilon-insensitive loss for regression problems with finite samples. To this end, we compare generalization performance of SVM regression (using proposed selection of epsilon-values) with regression using 'least-modulus' loss (epsilon=0) and standard squared loss. These comparisons indicate superior generalization performance of SVM regression under sparse sample settings, for various types of additive noise.
An overview on clustering methods
[EB/OL].(
Mastering the game of Go with deep neural networks and tree search
[J].DOI:10.1038/nature16961 [本文引用: 1]
Scalable deep reinforcement learning for vision-based robotic manipulation
[EB/OL]. (
Computer-assisted design of complex organic syntheses
[J].
Computational approaches streamlining drug discovery
[J].DOI:10.1038/s41586-023-05905-z [本文引用: 1]
机器学习在有机化学中的应用
[J].
DOI:10.6023/cjoc202006051
[本文引用: 1]
近年来,由于计算能力、大数据和算法的不断进步,人工智能(Artificial intelligence,AI)重新兴起,已成为诸多研究领域变革性发展背后的重要推动力.机器学习(Machine learning,ML)是人工智能一个重要的研究领域.随着化学信息学的发展,机器学习在化学领域展现出巨大的发展潜力,也为有机化学的发展带来了新的机遇.为帮助有机化学家了解这一新兴领域,对如何将机器学习策略应用于有机化学研究做简单介绍,同时,概括总结了机器学习在化合物性质预测、分子从头设计、化学反应预测、逆合成分析和智能合成机器方面的应用实例,分析讨论了当前机器学习在有机化学领域面临的挑战和难题.
Application of machine learning in organic chemistry
[J].
DOI:10.6023/cjoc202006051
[本文引用: 1]
Driven by nowadays’ computing power, big data technology as well as learning algorithm, artificial intelligence (AI) has gained trenmendous attentions and become a transformative approach in many research areas. One of the most extensively explored AI approaches in chemistry is (deep) machine learning, which provides new twists in the fields of organic chemistry. The workflow of machine learning (ML) study in organic chemistry is briefly introduced. Meanwhile, the application of ML in the accurate prediction of chemical properties, molecular <i>de novo</i> design, chemical reaction prediction, retrosynthetic analysis and artificial intelligence synthetic machine are also summarized. In the end, the current challenges in this field are analyzed and discussed.
Random forests
[J].DOI:10.1023/A:1010933404324 URL [本文引用: 1]
A unified machine-learning protocol for asymmetric catalysis as a proof of concept demonstration using asymmetric hydrogenation
[J].
DOI:10.1073/pnas.1916392117
PMID:31915295
[本文引用: 2]
Design of asymmetric catalysts generally involves time- and resource-intensive heuristic endeavors. In view of the steady increase in interest toward efficient catalytic asymmetric reactions and the rapid growth in the field of machine learning (ML) in recent years, we envisaged dovetailing these two important domains. We selected a set of quantum chemically derived molecular descriptors from five different asymmetric binaphthyl-derived catalyst families with the propensity to impact the enantioselectivity of asymmetric hydrogenation of alkenes and imines. The predictive power of the random forest (RF) built using the molecular parameters of a set of 368 substrate-catalyst combinations is found to be impressive, with a root-mean-square error (rmse) in the predicted enantiomeric excess (%) of about 8.4 ± 1.8 compared to the experimentally known values. The accuracy of RF is found to be superior to other ML methods such as convolutional neural network, decision tree, and eXtreme gradient boosting as well as stepwise linear regression. The proposed method is expected to provide a leap forward in the design of catalysts for asymmetric transformations.
Scikit-learn: Machine learning in python
[EB/OL]. (
Prediction of molecular electronic transitions using random forests
[J].
DOI:10.1021/acs.jcim.0c00698
PMID:33090804
[本文引用: 1]
Fluorescent molecules, fluorophores or dyes, play essential roles in bioimaging. Effective bioimaging requires fluorophores with diverse colors and high quantum yields for better resolution. An essential computational component to design novel dye molecules is an accurate model that predicts the electronic properties of molecules. Here, we present statistical machines that predict the excitation energies and associated oscillator strengths of a given molecule using the random forest algorithm. The excitation energies and oscillator strengths of a molecule are closely related to the emission spectrum and the quantum yields of fluorophores, respectively. In this study, we identified specific molecular substructures that induce high oscillator strengths of molecules. The results of our study are expected to serve as new design principles for designing novel fluorophores.
Predicting regioselectivity in radical C—H functionalization of heterocycles through machine learning
[J].DOI:10.1002/anie.v59.32 URL [本文引用: 1]
Predicting reaction performance in C—N cross-coupling using machine learning
[J].
DOI:10.1126/science.aar5169
URL
[本文引用: 2]
\n Chemists often discover reactions by applying catalysts to a series of simple compounds. Tweaking those reactions to tolerate more structural complexity in pharmaceutical research is time-consuming. Ahneman\n et al.\n report that machine learning can help. Using a high-throughput data set, they trained a random forest algorithm to predict which specific palladium catalysts would best tolerate isoxazoles (cyclic structures with an N–O bond) during C–N bond formation. The predictions also helped to guide analysis of the catalyst inhibition mechanism.\n
Enantioselectivity prediction of pallada-electrocatalysed C—H activation using transition state knowledge in machine learning
[J].DOI:10.1038/s44160-022-00233-y [本文引用: 2]
Recent advances in convolutional neural networks
[J].DOI:10.1016/j.patcog.2017.10.013 URL [本文引用: 1]
Convolutional neural network based on SMILES representation of compounds for detecting chemical motif
[J].DOI:10.1186/s12859-018-2523-5 [本文引用: 1]
AtomNet: A deep convolutional neural network for bioactivity prediction in structure-based drug discovery
[EB/OL].(
Modeling epoxidation of drug-like molecules with a deep machine learning network
[J].
DOI:10.1021/acscentsci.5b00131
PMID:27162970
[本文引用: 1]
Drug toxicity is frequently caused by electrophilic reactive metabolites that covalently bind to proteins. Epoxides comprise a large class of three-membered cyclic ethers. These molecules are electrophilic and typically highly reactive due to ring tension and polarized carbon-oxygen bonds. Epoxides are metabolites often formed by cytochromes P450 acting on aromatic or double bonds. The specific location on a molecule that undergoes epoxidation is its site of epoxidation (SOE). Identifying a molecule's SOE can aid in interpreting adverse events related to reactive metabolites and direct modification to prevent epoxidation for safer drugs. This study utilized a database of 702 epoxidation reactions to build a model that accurately predicted sites of epoxidation. The foundation for this model was an algorithm originally designed to model sites of cytochromes P450 metabolism (called XenoSite) that was recently applied to model the intrinsic reactivity of diverse molecules with glutathione. This modeling algorithm systematically and quantitatively summarizes the knowledge from hundreds of epoxidation reactions with a deep convolution network. This network makes predictions at both an atom and molecule level. The final epoxidation model constructed with this approach identified SOEs with 94.9% area under the curve (AUC) performance and separated epoxidized and non-epoxidized molecules with 79.3% AUC. Moreover, within epoxidized molecules, the model separated aromatic or double bond SOEs from all other aromatic or double bonds with AUCs of 92.5% and 95.1%, respectively. Finally, the model separated SOEs from sites of sp(2) hydroxylation with 83.2% AUC. Our model is the first of its kind and may be useful for the development of safer drugs. The epoxidation model is available at http://swami.wustl.edu/xenosite.
Site of reactivity models predict molecular reactivity of diverse chemicals with glutathione
[J].
DOI:10.1021/acs.chemrestox.5b00017
PMID:25742281
[本文引用: 1]
Drug toxicity is often caused by electrophilic reactive metabolites that covalently bind to proteins. Consequently, the quantitative strength of a molecule's reactivity with glutathione (GSH) is a frequently used indicator of its toxicity. Through cysteine, GSH (and proteins) scavenges reactive molecules to form conjugates in the body. GSH conjugates to specific atoms in reactive molecules: their sites of reactivity. The value of knowing a molecule's sites of reactivity is unexplored in the literature. This study tests the value of site of reactivity data that identifies the atoms within 1213 reactive molecules that conjugate to GSH and builds models to predict molecular reactivity with glutathione. An algorithm originally written to model sites of cytochrome P450 metabolism (called XenoSite) finds clear patterns in molecular structure that identify sites of reactivity within reactive molecules with 90.8% accuracy and separate reactive and unreactive molecules with 80.6% accuracy. Furthermore, the model output strongly correlates with quantitative GSH reactivity data in chemically diverse, external data sets. Site of reactivity data is nearly unstudied in the literature prior to our efforts, yet it contains a strong signal for reactivity that can be utilized to more accurately predict molecule reactivity and, eventually, toxicity.
Recognizing contamination fragment ions in liquid chromatography-tandem mass spectrometry data
[J].DOI:10.1021/jasms.0c00478 URL [本文引用: 1]
Fast acquisition of high-quality nuclear magnetic resonance pure shift spectroscopy via a deep neural network
[J].DOI:10.1021/acs.jpclett.2c00100 URL [本文引用: 3]
Geometric deep learning: Going beyond euclidean data
[J].
GNNExplainer: Generating explanations for graph neural networks
[EB/OL]. (
Gated graph sequence neural networks
[EB/OL]. (
Semi-supervised classification with graph convolutional networks
[EB/OL].(
A graph-convolutional neural network model for the prediction of chemical reactivity
[J].
DOI:10.1039/c8sc04228d
PMID:30746086
We present a supervised learning approach to predict the products of organic reactions given their reactants, reagents, and solvent(s). The prediction task is factored into two stages comparable to manual expert approaches: considering possible sites of reactivity and evaluating their relative likelihoods. By training on hundreds of thousands of reaction precedents covering a broad range of reaction types from the patent literature, the neural model makes informed predictions of chemical reactivity. The model predicts the major product correctly over 85% of the time requiring around 100 ms per example, a significantly higher accuracy than achieved by previous machine learning approaches, and performs on par with expert chemists with years of formal training. We gain additional insight into predictions the design of the neural model, revealing an understanding of chemistry qualitatively consistent with manual approaches.
Graph neural networks for predicting chemical reaction performance
[EB/OL]. (
Deep learning in chemistry
[J].
DOI:10.1021/acs.jcim.9b00266
PMID:31194543
Machine learning enables computers to address problems by learning from data. Deep learning is a type of machine learning that uses a hierarchical recombination of features to extract pertinent information and then learn the patterns represented in the data. Over the last eight years, its abilities have increasingly been applied to a wide variety of chemical challenges, from improving computational chemistry to drug and materials design and even synthesis planning. This review aims to explain the concepts of deep learning to chemists from any background and follows this with an overview of the diverse applications demonstrated in the literature. We hope that this will empower the broader chemical community to engage with this burgeoning field and foster the growing movement of deep learning accelerated chemistry.
Rapid and accurate prediction of pKa values of C—H acids using graph convolutional neural networks
[J].
BonDNet: A graph neural network for the prediction of bond dissociation energies for charged molecules
[J].Prediction of bond dissociation energies for charged molecules with a graph neural network enabled by global molecular features and reaction difference features between products and reactants.
Deep learning of activation energies
[J].DOI:10.1021/acs.jpclett.0c00500 URL
Prediction of higher-selectivity catalysts by computer-driven workflow and machine learning
[J].
DOI:10.1126/science.aau5631
URL
[本文引用: 3]
\n Asymmetric catalysis is widely used in chemical research and manufacturing to access just one of two possible mirror-image products. Nonetheless, the process of tuning catalyst structure to optimize selectivity is still largely empirical. Zahrt\n et al.\n present a framework for more efficient, predictive optimization. As a proof of principle, they focused on a known coupling reaction of imines and thiols catalyzed by chiral phosphoric acid compounds. By modeling multiple conformations of more than 800 prospective catalysts, and then training machine-learning algorithms on a subset of experimental results, they achieved highly accurate predictions of enantioselectivities.\n
Planning chemical syntheses with deep neural networks and symbolic AI
[J].DOI:10.1038/nature25978 URL [本文引用: 3]
Imaging through glass diffusers using densely connected convolutional networks
[J].DOI:10.1364/OPTICA.5.000803 URL [本文引用: 1]
MoleculeNet: A benchmark for molecular machine learning
[J].DOI:10.1039/C7SC02664A URL [本文引用: 1]
Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17
[J].
DOI:10.1021/ci300415d
PMID:23088335
[本文引用: 1]
Drug molecules consist of a few tens of atoms connected by covalent bonds. How many such molecules are possible in total and what is their structure? This question is of pressing interest in medicinal chemistry to help solve the problems of drug potency, selectivity, and toxicity and reduce attrition rates by pointing to new molecular series. To better define the unknown chemical space, we have enumerated 166.4 billion molecules of up to 17 atoms of C, N, O, S, and halogens forming the chemical universe database GDB-17, covering a size range containing many drugs and typical for lead compounds. GDB-17 contains millions of isomers of known drugs, including analogs with high shape similarity to the parent drug. Compared to known molecules in PubChem, GDB-17 molecules are much richer in nonaromatic heterocycles, quaternary centers, and stereoisomers, densely populate the third dimension in shape space, and represent many more scaffold types.
SchNetPack: A deep learning toolbox for atomistic systems
[J].
DOI:10.1021/acs.jctc.8b00908
PMID:30481453
[本文引用: 1]
SchNetPack is a toolbox for the development and application of deep neural networks that predict potential energy surfaces and other quantum-chemical properties of molecules and materials. It contains basic building blocks of atomistic neural networks, manages their training, and provides simple access to common benchmark datasets. This allows for an easy implementation and evaluation of new models. For now, SchNetPack includes implementations of (weighted) atom-centered symmetry functions and the deep tensor neural network SchNet, as well as ready-to-use scripts that allow one to train these models on molecule and material datasets. Based on the PyTorch deep learning framework, SchNetPack allows one to efficiently apply the neural networks to large datasets with millions of reference calculations, as well as parallelize the model across multiple GPUs. Finally, SchNetPack provides an interface to the Atomic Simulation Environment in order to make trained models easily accessible to researchers that are not yet familiar with neural networks.
Evaluation guidelines for machine learning tools in the chemical sciences
[J].
DOI:10.1038/s41570-022-00391-9
PMID:37117429
[本文引用: 1]
Machine learning (ML) promises to tackle the grand challenges in chemistry and speed up the generation, improvement and/or ordering of research hypotheses. Despite the overarching applicability of ML workflows, one usually finds diverse evaluation study designs. The current heterogeneity in evaluation techniques and metrics leads to difficulty in (or the impossibility of) comparing and assessing the relevance of new algorithms. Ultimately, this may delay the digitalization of chemistry at scale and confuse method developers, experimentalists, reviewers and journal editors. In this Perspective, we critically discuss a set of method development and evaluation guidelines for different types of ML-based publications, emphasizing supervised learning. We provide a diverse collection of examples from various authors and disciplines in chemistry. While taking into account varying accessibility across research groups, our recommendations focus on reporting completeness and standardizing comparisons between tools. We aim to further contribute to improved ML transparency and credibility by suggesting a checklist of retro-/prospective tests and dissecting their importance. We envisage that the wide adoption and continuous update of best practices will encourage an informed use of ML on real-world problems related to the chemical sciences.© 2022. Springer Nature Limited.
Crowdsourced mapping of unexplored target space of kinase inhibitors
[J].
DOI:10.1038/s41467-020-20314-w
[本文引用: 1]
The ice arches that usually develop at the northern and southern ends of Nares Strait play an important role in modulating the export of Arctic Ocean multi-year sea ice. The Arctic Ocean is evolving towards an ice pack that is younger, thinner, and more mobile and the fate of its multi-year ice is becoming of increasing interest. Here, we use sea ice motion retrievals from Sentinel-1 imagery to report on the recent behavior of these ice arches and the associated ice fluxes. We show that the duration of arch formation has decreased over the past 20 years, while the ice area and volume fluxes along Nares Strait have both increased. These results suggest that a transition is underway towards a state where the formation of these arches will become atypical with a concomitant increase in the export of multi-year ice accelerating the transition towards a younger and thinner Arctic ice pack.
Machine learning approach for prediction of reaction yield with simulated catalyst parameters
[J].DOI:10.1246/cl.171130 URL [本文引用: 1]
Enantioselective dehydrogenative heck arylations of trisubstituted alkenes with indoles to construct quaternary stereocenters
[J].
DOI:10.1021/jacs.5b11335
PMID:26624236
[本文引用: 1]
An enantioselective, intermolecular dehydrogenative Heck arylation of trisubstituted alkenes to construct remote quaternary stereocenters has been developed. Using a new chiral pyridine oxazoline ligand, good to high enantioselectivity is achieved for various combinations of indole derivatives and trisubstituted alkenes. However, some combinations of substrates led to lower enantioselectivity, which provided the impetus to use structure enantioselectivity correlations to design a better performing ligand.
Quantifying structural effects of amino acid ligands in Pd(II)-catalyzed enantioselective C—H functionalization reactions
[J].DOI:10.1021/acs.organomet.7b00751 URL [本文引用: 1]
Developing comprehensive computational parameter sets to describe the performance of pyridine-oxazoline and related ligands
[J].DOI:10.1021/acscatal.7b00739 URL [本文引用: 1]
Design of experimental conditions with machine learning for collaborative organic synthesis reactions using transition-metal catalysts
[J].
DOI:10.1021/acsomega.1c04826
PMID:34693179
[本文引用: 1]
To improve product yields in synthetic reactions, it is important to use appropriate catalysts. In this study, we used machine learning to design catalysts for a reaction system in which both Buchwald-Hartwig-type and Suzuki-Miyaura-type cross-coupling reactions proceed simultaneously. First, using an existing dataset, yield prediction models were constructed with machine learning between experimental conditions, including the substrate and catalyst and the yields of the two products. Seven methods for calculating both the substrate and catalyst descriptors were proposed, and the predictive ability of the yield prediction models was discussed in terms of the descriptors and machine learning methods. Then, the constructed models were used to predict the compound yields for new combinations of substrates and catalysts, and the predictions were experimentally validated with high reproducibility, confirming that machine learning can predict yields from experimental conditions with high accuracy. In addition, to design catalysts that will improve the yields in our dataset, we added datasets collected from scientific papers and designed catalyst ligands. The proposed catalyst candidates were tested in actual synthetic experiments, and the experimental results exceeded the existing yields.© 2021 The Authors. Published by American Chemical Society.
Prediction of organic reaction outcomes using machine learning
[J].
DOI:10.1021/acscentsci.7b00064
PMID:28573205
[本文引用: 1]
Computer assistance in synthesis design has existed for over 40 years, yet retrosynthesis planning software has struggled to achieve widespread adoption. One critical challenge in developing high-quality pathway suggestions is that proposed reaction steps often fail when attempted in the laboratory, despite initially seeming viable. The true measure of success for any synthesis program is whether the predicted outcome matches what is observed experimentally. We report a model framework for anticipating reaction outcomes that combines the traditional use of reaction templates with the flexibility in pattern recognition afforded by neural networks. Using 15 000 experimental reaction records from granted United States patents, a model is trained to select the major (recorded) product by ranking a self-generated list of candidates where one candidate is known to be the major product. Candidate reactions are represented using a unique edit-based representation that emphasizes the fundamental transformation from reactants to products, rather than the constituent molecules' overall structures. In a 5-fold cross-validation, the trained model assigns the major product rank 1 in 71.8% of cases, rank ≤3 in 86.7% of cases, and rank ≤5 in 90.8% of cases.
A machine learning approach for predicting the nucleophilicity of organic molecules
[J].
DOI:10.1039/d1cp05072a
PMID:34986215
[本文引用: 1]
Nucleophilicity provides important information about the chemical reactivity of organic molecules. Experimental determination of the nucleophilicity parameter is a tedious and resource-intensive approach. Herein, we present a novel machine learning protocol that uses key structural descriptors to predict the nucleophilicities of organic molecules, which agree well with the experimental values. A data driven approach was used where quantum mechanical molecular and thermodynamic descriptors from a wide range of structurally diverse nucleophiles and relevant solvents were extracted and modelled using advanced algorithms against the experimentally available nucleophilicity values. Despite the structural diversity of nucleophiles, we are able to achieve statistically robust models with a high predictive power using tree-based and neural network algorithms trained on an in-house developed unique dataset consisting of 752 nucleophilicity values and 27 molecular descriptors.
Random forest models to predict aqueous solubility
[J].
DOI:10.1021/ci060164k
PMID:17238260
[本文引用: 1]
Random Forest regression (RF), Partial-Least-Squares (PLS) regression, Support Vector Machines (SVM), and Artificial Neural Networks (ANN) were used to develop QSPR models for the prediction of aqueous solubility, based on experimental data for 988 organic molecules. The Random Forest regression model predicted aqueous solubility more accurately than those created by PLS, SVM, and ANN and offered methods for automatic descriptor selection, an assessment of descriptor importance, and an in-parallel measure of predictive ability, all of which serve to recommend its use. The prediction of log molar solubility for an external test set of 330 molecules that are solid at 25 degrees C gave an r2 = 0.89 and RMSE = 0.69 log S units. For a standard data set selected from the literature, the model performed well with respect to other documented methods. Finally, the diversity of the training and test sets are compared to the chemical space occupied by molecules in the MDL drug data report, on the basis of molecular descriptors selected by the regression analysis.
Machine learning prediction of UV-Vis spectra features of organic compounds related to photoreactive potential
[J].
DOI:10.1038/s41598-020-79139-8
[本文引用: 1]
Our previous study demonstrated increased expression of Heat shock protein (Hsp) 90 in the skin of patients with systemic sclerosis (SSc). We aimed to evaluate plasma Hsp90 in SSc and characterize its association with SSc-related features. Ninety-two SSc patients and 92 age-/sex-matched healthy controls were recruited for the cross-sectional analysis. The longitudinal analysis comprised 30 patients with SSc associated interstitial lung disease (ILD) routinely treated with cyclophosphamide. Hsp90 was increased in SSc compared to healthy controls. Hsp90 correlated positively with C-reactive protein and negatively with pulmonary function tests: forced vital capacity and diffusing capacity for carbon monoxide (DLCO). In patients with diffuse cutaneous (dc) SSc, Hsp90 positively correlated with the modified Rodnan skin score. In SSc-ILD patients treated with cyclophosphamide, no differences in Hsp90 were found between baseline and after 1, 6, or 12 months of therapy. However, baseline Hsp90 predicts the 12-month change in DLCO. This study shows that Hsp90 plasma levels are increased in SSc patients compared to age-/sex-matched healthy controls. Elevated Hsp90 in SSc is associated with increased inflammatory activity, worse lung functions, and in dcSSc, with the extent of skin involvement. Baseline plasma Hsp90 predicts the 12-month change in DLCO in SSc-ILD patients treated with cyclophosphamide.
Machine learning for predicting product distributions in catalytic regioselective reactions
[J].
DOI:10.1039/c8cp03141j
PMID:29967920
[本文引用: 1]
Gaining predictable control over various forms of selectivities, such as enantio- and/or regio-selectivities, has been a long-standing goal in chemical catalysis. Although a number of factors such as the molecular features of the reactants and catalysts, as well as the reaction conditions, can influence the outcome of a reaction, it is not quite conspicuous as to what combinations of these parameters would offer a desired form of selectivity. We use machine learning tools, such as the neural network (NN), decision tree (DT), logistic regression (LR) and Random forest algorithms, to (a) analyze the outcome of an important catalytic regio-selective difluorination reaction of alkenes, and (b) decipher the complex interplay of various molecular parameters and their non-linear dependencies. The connection between what features of alkenes will yield 1,1-difluorination and how subtle changes would steer the reaction to 1,2-difluorination under identical conditions is enunciated. The NN was able to accurately predict whether a given alkene would yield a 1,1- or 1,2-difluorinated product. A combination of DT and the random forest classifier offered important chemical insights, which could be used in making a more rational choice of the reactant alkene for the desired regioisomeric product. The results could have far reaching implications in predicting which regioisomer is likely to be formed under a given set of conditions, and thus this technique is capable of expediting the development of catalytic transformations.
Structure-based classification of chemical reactions without assignment of reaction centers
[J].DOI:10.1021/ci0502707 URL [本文引用: 1]
Bayesian reaction optimization as a tool for chemical synthesis
[J].DOI:10.1038/s41586-021-03213-y [本文引用: 1]
A Bayesian graph convolutional network for reliable prediction of molecular properties with uncertainty quantification
[J].
DOI:10.1039/c9sc01992h
PMID:31803423
[本文引用: 1]
Deep neural networks have been increasingly used in various chemical fields. In the nature of a data-driven approach, their performance strongly depends on data used in training. Therefore, models developed in data-deficient situations can cause highly uncertain predictions, leading to vulnerable decision making. Here, we show that Bayesian inference enables more reliable prediction with quantitative uncertainty analysis. Decomposition of the predictive uncertainty into model- and data-driven uncertainties allows us to elucidate the source of errors for further improvements. For molecular applications, we devised a Bayesian graph convolutional network (GCN) and evaluated its performance for molecular property predictions. Our study on the classification problem of bio-activity and toxicity shows that the confidence of prediction can be quantified in terms of the predictive uncertainty, leading to more accurate virtual screening of drug candidates than standard GCNs. The result of log prediction illustrates that data noise affects the data-driven uncertainty more significantly than the model-driven one. Based on this finding, we could identify artefacts that arose from quantum mechanical calculations in the Harvard Clean Energy Project dataset. Consequently, the Bayesian GCN is critical for molecular applications under data-deficient conditions.This journal is © The Royal Society of Chemistry 2019.
Bayesian learning of chemisorption for bridging the complexity of electronic descriptors
[J].
DOI:10.1038/s41467-019-13993-7
[本文引用: 1]
Stimulated cells and cancer cells have widespread shortening of mRNA 3’-untranslated regions (3’UTRs) and switches to shorter mRNA isoforms due to usage of more proximal polyadenylation signals (PASs) in introns and last exons. U1 snRNP (U1), vertebrates’ most abundant non-coding (spliceosomal) small nuclear RNA, silences proximal PASs and its inhibition with antisense morpholino oligonucleotides (U1 AMO) triggers widespread premature transcription termination and mRNA shortening. Here we show that low U1 AMO doses increase cancer cells’ migration and invasion in vitro by up to 500%, whereas U1 over-expression has the opposite effect. In addition to 3’UTR length, numerous transcriptome changes that could contribute to this phenotype are observed, including alternative splicing, and mRNA expression levels of proto-oncogenes and tumor suppressors. These findings reveal an unexpected role for U1 homeostasis (available U1 relative to transcription) in oncogenic and activated cell states, and suggest U1 as a potential target for their modulation.
Optimizing chemical reactions with deep reinforcement learning
[J].
DOI:10.1021/acscentsci.7b00492
PMID:29296675
[本文引用: 1]
Deep reinforcement learning was employed to optimize chemical reactions. Our model iteratively records the results of a chemical reaction and chooses new experimental conditions to improve the reaction outcome. This model outperformed a state-of-the-art blackbox optimization algorithm by using 71% fewer steps on both simulations and real reactions. Furthermore, we introduced an efficient exploration strategy by drawing the reaction conditions from certain probability distributions, which resulted in an improvement on regret from 0.062 to 0.039 compared with a deterministic policy. Combining the efficient exploration policy with accelerated microdroplet reactions, optimal reaction conditions were determined in 30 min for the four reactions considered, and a better understanding of the factors that control microdroplet reactions was reached. Moreover, our model showed a better performance after training on reactions with similar or even dissimilar underlying mechanisms, which demonstrates its learning ability.
Reinforcement learning for molecular design guided by quantum mechanics
[EB/OL]. (
Deep reinforcement learning forde novo drug design
[J].
DOI:10.1126/sciadv.aap7885
URL
[本文引用: 1]
We introduce an artificial intelligence approach to de novo design of molecules with desired physical or biological properties.
De novo drug design using reinforcement learning with graph-based deep generative models
[J].
DOI:10.1021/acs.jcim.2c00838
PMID:36219571
[本文引用: 1]
Machine learning provides effective computational tools for exploring the chemical space via deep generative models. Here, we propose a new reinforcement learning scheme to fine-tune graph-based deep generative models for molecular design tasks. We show how our computational framework can successfully guide a pretrained generative model toward the generation of molecules with a specific property profile, even when such molecules are not present in the training set and unlikely to be generated by the pretrained model. We explored the following tasks: generating molecules of decreasing/increasing size, increasing drug-likeness, and increasing bioactivity. Using the proposed approach, we achieve a model which generates diverse compounds with predicted DRD2 activity for 95% of sampled molecules, outperforming previously reported methods on this metric.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}