
收稿日期: 2023-03-06
修回日期: 2023-05-12
录用日期: 2023-05-16
网络出版日期: 2023-05-24
基金资助
国家自然科学基金面上项目(22071147)
Application of Machine Learning in Chemical Synthesis and Characterization
Received date: 2023-03-06
Revised date: 2023-05-12
Accepted date: 2023-05-16
Online published: 2023-05-24
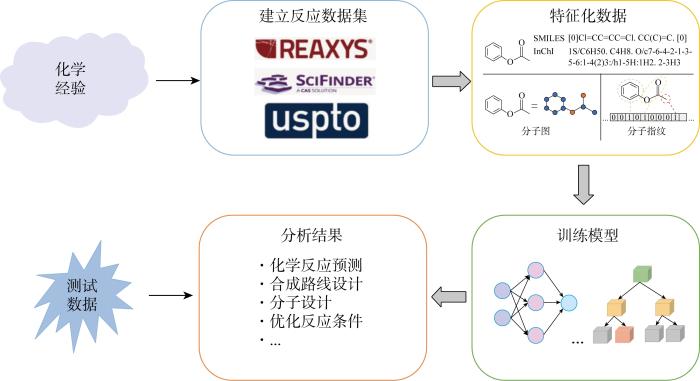
自动化化学合成是化学领域长期追求的目标之一.近年来,机器学习的出现使得实现这一目标有了可能.以数据驱动为核心的机器学习借助计算机学习海量化学数据中的信息,寻找信息之间的客观联系和规律,根据已有规律和信息训练生成模型,借助模型预测分析需解决的实际问题.机器学习因其出色的计算预测能力,帮助化学工作者快速高效解决化学合成问题,加快研究进程.机器学习的出现和发展对化学合成及表征领域展示出强大的研究助力作用,但目前并不存在通用性极强的机器学习模型,化学工作者仍需根据实际情况选择不同模型进行训练学习.从监督学习、无监督学习、半监督学习、强化学习等机器学习的角度,向化学工作者展示常见学习方法在化学合成及表征中应用的最佳案例,帮助其利用机器学习知识进一步拓宽研究思路.
孙婕 , 李子昊 , 张书宇 . 机器学习在化学合成及表征中的应用[J]. 上海交通大学学报, 2023 , 57(10) : 1231 -1244 . DOI: 10.16183/j.cnki.jsjtu.2023.078
Automated chemical synthesis is one of the long-term goals pursued in the field of chemistry. In recent years, the advent of machine learning (ML) has made it possible to achieve this goal. Data-driven ML uses computers to learn relative information in massive chemical data, find objective connections between information, train models by using objective connections, and analyze the actual problems which can be solved according to these models. With its excellent computational prediction capabilities, ML helps chemists solve chemical synthesis problems quickly and efficiently and accelerate the research process. The emergence and development of ML has shown a strong research assistance in the field of chemical synthesis and characterization. However, there is no highly versatile ML model at present, and chemists still need to choose different models for training and learning according to actual situations. This paper aims to show chemists the best cases of common learning methods in chemical synthesis and characterization from the perspective of ML, such as supervised learning, unsupervised learning, semi-supervised learning, reinforcement learning, etc., and help them use ML knowledge to further broaden their research ideas.
| [1] | JORDAN M I, MITCHELL T M. Machine learning: Trends, perspectives, and prospects[J]. Science, 2015, 349(6245): 255-260. |
| [2] | BUTLER K T, DAVIES D W, CARTWRIGHT H, et al. Machine learning for molecular and materials science[J]. Nature, 2018, 559(7715): 547-555. |
| [3] | WANG X R, LI Y Q, QIU J Z, et al. RetroPrime: A Diverse, plausible and Transformer-based method for Single-Step retrosynthesis predictions[J]. Chemical Engineering Journal, 2021, 420: 129845. |
| [4] | SCHWALLER P, LAINO T, GAUDIN T, et al. Molecular transformer: A model for uncertainty-calibrated chemical reaction prediction[J]. ACS Central Science, 2019, 5(9): 1572-1583. |
| [5] | WEI J N, DUVENAUD D, ASPURU-GUZIK A. Neural networks for the prediction of organic chemistry reactions[J]. ACS Central Science, 2016, 2(10): 725-732. |
| [6] | NAM J, KIM J. Linking the neural machine translation and the prediction of organic chemistry reactions[EB/OL].(2016-12-29) [2022-03-05].https://arxiv.org/abs/1612.09529. |
| [7] | LIU B W, RAMSUNDAR B, KAWTHEKAR P, et al. Retrosynthetic reaction prediction using neural sequence-to-sequence models[J]. ACS Central Science, 2017, 3(10): 1103-1113. |
| [8] | SEGLER M, PREU? M, WALLER M P. Towards "AlphaChem": Chemical synthesis planning with tree search and deep neural network policies[EB/OL]. (2017-01-31)[2022-03-05]. https://arxiv.org/abs/1702.00020. |
| [9] | SANCHEZ-LENGELING B, OUTEIRAL C, GUIMARAES G L, et al. Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC)[EB/OL]. (2017-08-18)[2022-03-05]. https://chemrxiv.org/engage/chemrxiv/article-details/60c73d91702a9beea7189bc2. |
| [10] | SARKER I H, HOQUE M M, UDDIN M K, et al. Mobile data science and intelligent apps: Concepts, AI-based modeling and research directions[J]. Mobile Networks and Applications, 2021, 26(1): 285-303. |
| [11] | SARKER I H, KAYES A S M, BADSHA S, et al. Cybersecurity data science: An overview from machine learning perspective[J]. Journal of Big Data, 2020, 7(1): 41. |
| [12] | WARR W A. A short review of chemical reaction database systems, computer-aided synthesis design, reaction prediction and synthetic feasibility[J]. Molecular Informatics, 2014, 33(6/7): 469-476. |
| [13] | WEININGER D. SMILES, a chemical language and information system. 1. introduction to methodology and encoding rules[J]. Journal of Chemical Information & Computer Sciences, 1988, 28(1): 31-36. |
| [14] | JELIAZKOVA N, KOCHEV N. AMBIT-SMARTS: Efficient searching of chemical structures and fragments[J]. Molecular Informatics, 2011, 30(8): 707-720. |
| [15] | LI Z H, LI Q Z, BAI H Y, et al. Synthetic strategies and mechanistic studies of axially chiral styrenes[J]. Chem Catalysis, 2023, 3: 100594. |
| [16] | TOMBERG A, JOHANSSON M J, NORRBY P O. A predictive tool for electrophilic aromatic substitutions using machine learning[J]. The Journal of Organic Chemistry, 2019, 84(8): 4695-4703. |
| [17] | LI Q Z, LI Z H, KANG J C, et al. Ni-catalyzed, enantioselective three-component radical relayed reductive coupling of alkynes: Synthesis of axially chiral styrenes[J]. Chem Catalysis, 2022, 2(11): 3185-3195. |
| [18] | CHEN C, ZHANG S Y, LI S X, et al. Electroreductive fluoroalkylative heteroarylation of unactivated alkenes via an unconventional remote heteroaryl migration[J]. Cell Reports Physical Science, 2023, 4(5): 101385. |
| [19] | SARKER I H. Machine learning: Algorithms, real-world applications and research directions[J]. SN Computer Science, 2021, 2(3): 160. |
| [20] | HAN J, PEI J, TONG H. Data mining: Concepts and techniques[M]. San Mateo: Morgan Kaufmann, 2022. |
| [21] | WANG H F, HU D J. Comparison of SVM and LS-SVM for regression[C]// 2005 International Conference on Neural Networks and Brain. Beijing, China: IEEE, 2005: 279-283. |
| [22] | CHERKASSKY V, MA Y Q. Practical selection of SVM parameters and noise estimation for SVM regression[J]. Neural Networks, 2004, 17(1): 113-126. |
| [23] | CORTES C, VAPNIK V. Support-vector networks[J]. Machine Learning, 1995, 20(3): 273-297. |
| [24] | HAMERLY G, ELKAN C. Learning the k in k-means[C]// Proceedings of the 16th International Conference on Neural Information Processing Systems. Cambridge, MA, USA: MIT Press, 2003: 281-288. |
| [25] | MONTGOMERY D C, PECK E A, VINING G G. Introduction to linear regression analysis[M]. 6th ed. Hoboken: Wiley, 2021. |
| [26] | MAHALAXMI K V K, REKHA K S. Comparison of logistic regression and artificial neural network for modelling credit card data set with the identification of precise fraudulent[C]//2022 International Conference on Business Analytics for Technology and Security. Dubai: IEEE, 2022: 1-5. |
| [27] | MADHULATHA T S. An overview on clustering methods[EB/OL].(2012-05-05)[2022-03-05]. https://arxiv.org/abs/1205.1117. |
| [28] | LAPAN M. Deep reinforcement learning hands-on:Apply modern RL methods, with deep Q-networks, value iteration, policy gradients, TRPO, AlphaGo Zero and more[M]. Mumbai: Packt Publishing, 2018. |
| [29] | SILVER D, HUANG A, MADDISON C J, et al. Mastering the game of Go with deep neural networks and tree search[J]. Nature, 2016, 529(7587): 484-489. |
| [30] | KALASHNIKOV D, IRPAN A, PASTOR P, et al. Scalable deep reinforcement learning for vision-based robotic manipulation[EB/OL]. (2018-06-27)[2022-03-05].https://arxiv.org/abs/1806.10293v3. |
| [31] | COREY E J, WIPKE W T. Computer-assisted design of complex organic syntheses[J]. Science, 1969, 166(3902): 178-192. |
| [32] | SADYBEKOV A V, KATRITCH V. Computational approaches streamlining drug discovery[J]. Nature, 2023, 616(7958): 673-685. |
| [33] | 刘伊迪, 杨骐, 李遥, 等. 机器学习在有机化学中的应用[J]. 有机化学, 2020, 40(11): 3812-3827. |
| [33] | LIU Yidi, YANG Qi, LI Yao, et al. Application of machine learning in organic chemistry[J]. Chinese Journal of Organic Chemistry, 2020, 40(11): 3812-3827. |
| [34] | BREIMAN L. Random forests[J]. Machine Learning, 2001, 45(1): 5-32. |
| [35] | SINGH S, PAREEK M, CHANGOTRA A, et al. A unified machine-learning protocol for asymmetric catalysis as a proof of concept demonstration using asymmetric hydrogenation[J]. Proceedings of the National Academy of Sciences of the United States of America, 2020, 117(3): 1339-1345. |
| [36] | PEDREGOSA F, VAROQUAUX G, GRAMFORT A, et al. Scikit-learn: Machine learning in python[EB/OL]. (2012-01-02)[2022-03-05].https://arxiv.org/abs/1201.0490. |
| [37] | KANG B, SEOK C, LEE J Y. Prediction of molecular electronic transitions using random forests[J]. Journal of Chemical Information and Modeling, 2020, 60(12): 5984-5994. |
| [38] | LI X, ZHANG S Q, XU L C, et al. Predicting regioselectivity in radical C—H functionalization of heterocycles through machine learning[J]. Angewandte Chemie International Edition, 2020, 59(32): 13253-13259. |
| [39] | AHNEMAN D T, ESTRADA J G, LIN S S, et al. Predicting reaction performance in C—N cross-coupling using machine learning[J]. Science, 2018, 360(6385): 186-190. |
| [40] | XU L C, FREY J, HOU X Y, et al. Enantioselectivity prediction of pallada-electrocatalysed C—H activation using transition state knowledge in machine learning[J]. Nature Synthesis, 2023, 2(4): 321-330. |
| [41] | LECUN Y, BOSER B, DENKER J, et al. Handwritten digit recognition with a back-propagation network[C]//Proceedings of the 2nd International Conference on Neural Information Processing Systems. Cambridge, MA, USA: MIT Press, 1989: 396-404. |
| [42] | LECUN Y, BOTTOU L, ORR G B, et al. Efficient BackProp[M]//Neural networks:Tricks of the trade. Berlin, Heidelberg: Springer, 2012: 9-48. |
| [43] | NAIR V, HINTON G E. Rectified linear units improve restricted boltzmann machines[C]//Proceedings of the 27th International Conference on International Conference on Machine Learning. Madison, WI, USA: Omnipress, 2010: 807-814. |
| [44] | WANG T, WU D J, COATES A, et al. End-to-end text recognition with convolutional neural networks[C]// Proceedings of the 21 st International Conference on Pattern Recognition. Tsukuba, Japan: IEEE, 2012: 3304-3308. |
| [45] | GU J X, WANG Z H, KUEN J, et al. Recent advances in convolutional neural networks[J]. Pattern Recognition, 2018, 77: 354-377. |
| [46] | ZINKEVICH M A, WEIMER M, SMOLA A, et al. Parallelized stochastic gradient descent[C]// Proceedings of the 23rd International Conference on Neural Information Processing Systems. Red Hook, NY, USA: Curran Associates Inc., 2010: 2595-2603. |
| [47] | HIROHARA M, SAITO Y, KODA Y, et al. Convolutional neural network based on SMILES representation of compounds for detecting chemical motif[J]. BMC Bioinformatics, 2018, 19(Sup.19): 526. |
| [48] | WALLACH I, DZAMBA M, HEIFETS A. AtomNet: A deep convolutional neural network for bioactivity prediction in structure-based drug discovery[EB/OL].(2015-10-10)[2022-03-05].https://arxiv.org/abs/1510.02855. |
| [49] | HUGHES T B, MILLER G P, SWAMIDASS S J. Modeling epoxidation of drug-like molecules with a deep machine learning network[J]. ACS Central Science, 2015, 1(4): 168-180. |
| [50] | HUGHES T B, MILLER G P, SWAMIDASS S J. Site of reactivity models predict molecular reactivity of diverse chemicals with glutathione[J]. Chemical Research in Toxicology, 2015, 28(4): 797-809. |
| [51] | XING S P, YU H X, LIU M, et al. Recognizing contamination fragment ions in liquid chromatography-tandem mass spectrometry data[J]. Journal of the American Society for Mass Spectrometry, 2021, 32(9): 2296-2305. |
| [52] | ZHENG X X, YANG Z X, YANG C, et al. Fast acquisition of high-quality nuclear magnetic resonance pure shift spectroscopy via a deep neural network[J]. The Journal of Physical Chemistry Letters, 2022, 13(9): 2101-2106. |
| [53] | BRONSTEIN M M, BRUNA J, LECUN Y, et al. Geometric deep learning: Going beyond euclidean data[J]. IEEE Signal Processing Magazine, 2017, 34(4): 18-42. |
| [54] | YING R, BOURGEOIS D, YOU J X, et al. GNNExplainer: Generating explanations for graph neural networks[EB/OL]. (2019-03-10)[2022-03-05]. https://arxiv.org/abs/1903.03894. |
| [55] | LI Y J, TARLOW D, BROCKSCHMIDT M, et al. Gated graph sequence neural networks[EB/OL]. (2015-11-17)[2022-03-05].https://arxiv.org/abs/1511.05493. |
| [56] | KIPF T N, WELLING M. Semi-supervised classification with graph convolutional networks[EB/OL].(2016-09-09)[2022-03-05].https://arxiv.org/abs/1609.02907. |
| [57] | DUVENAUD D, MACLAURIN D, AGUILERA-IPARRAGUIRRE J, et al. Convolutional networks on graphs for learning molecular fingerprints[C]//Proceedings of the 28th International Conference on Neural Information Processing Systems-Volume 2. Montreal, Canada: ACM, 2015: 2224-2232. |
| [58] | COLEY C, JIN W G, ROGERS L, et al. A graph-convolutional neural network model for the prediction of chemical reactivity[J]. Chemical Science, 2019, 10(2): 370-377. |
| [59] | SAEBI M, NAN B, HERR J E, et al. Graph neural networks for predicting chemical reaction performance[EB/OL]. (2021-05-01)[2022-03-05]. https://www.researchgate.net/publication/351635791_Graph_Neural_Networks_for_Predicting_Chemical_Reaction_Performance. |
| [60] | MATER A C, COOTE M L. Deep learning in chemistry[J]. Journal of Chemical Information and Modeling, 2019, 59(6): 2545-2559. |
| [61] | ROSZAK R, BEKER W, MOLGA K, et al. Rapid and accurate prediction of pKa values of C—H acids using graph convolutional neural networks[J]. Journal of the American Chemical Society, 2019, 141(43): 17142-17149. |
| [62] | WEN M J, BLAU S M, SPOTTE-SMITH E W C, et al. BonDNet: A graph neural network for the prediction of bond dissociation energies for charged molecules[J]. Chemical Science, 2021, 12(5): 1858-1868. |
| [63] | GRAMBOW C A, PATTANAIK L, GREEN W H. Deep learning of activation energies[J]. The Journal of Physical Chemistry Letters, 2020, 11(8): 2992-2997. |
| [64] | ZAHRT A F, HENLE J J, ROSE B T, et al. Prediction of higher-selectivity catalysts by computer-driven workflow and machine learning[J]. Science, 2019, 363(6424): eaau5631. |
| [65] | VAN OTTERLO M, WIERING M. Reinforcement learning and Markov decision processes[M]//Adaptation, learning, and optimization. Berlin, Heidelberg: Springer, 2012: 3-42. |
| [66] | SEGLER M H S, PREUSS M, WALLER M P. Planning chemical syntheses with deep neural networks and symbolic AI[J]. Nature, 2018, 555(7698): 604-610. |
| [67] | LI S, DENG M, LEE J, et al. Imaging through glass diffusers using densely connected convolutional networks[J]. Optica, 2018, 5(7): 803. |
| [68] | WU Z Q, RAMSUNDAR B, FEINBERG E N, et al. MoleculeNet: A benchmark for molecular machine learning[J]. Chemical Science, 2017, 9(2): 513-530. |
| [69] | RUDDIGKEIT L, VAN DEURSEN R, BLUM L C, et al. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17[J]. Journal of Chemical Information and Modeling, 2012, 52(11): 2864-2875. |
| [70] | SCHüTT K T, KESSEL P, GASTEGGER M, et al. SchNetPack: A deep learning toolbox for atomistic systems[J]. Journal of Chemical Theory and Computation, 2019, 15(1): 448-455. |
| [71] | BENDER A, SCHNEIDER N, SEGLER M, et al. Evaluation guidelines for machine learning tools in the chemical sciences[J]. Nature Reviews Chemistry, 2022, 6(6): 428-442. |
| [72] | CICHO?SKA A, RAVIKUMAR B, ALLAWAY R J, et al. Crowdsourced mapping of unexplored target space of kinase inhibitors[J]. Nature Communications, 2021, 12(1): 1-18. |
| [73] | YADA A, NAGATA K, ANDO Y, et al. Machine learning approach for prediction of reaction yield with simulated catalyst parameters[J]. Chemistry Letters, 2018, 47(3): 284-287. |
| [74] | ZHANG C, SANTIAGO C B, CRAWFORD J M, et al. Enantioselective dehydrogenative heck arylations of trisubstituted alkenes with indoles to construct quaternary stereocenters[J]. Journal of the American Chemical Society, 2015, 137(50): 15668-15671. |
| [75] | PARK Y, NIEMEYER Z L, YU J Q, et al. Quantifying structural effects of amino acid ligands in Pd(II)-catalyzed enantioselective C—H functionalization reactions[J]. Organometallics, 2018, 37(2): 203-210. |
| [76] | GUO J Y, MINKO Y, SANTIAGO C B, et al. Developing comprehensive computational parameter sets to describe the performance of pyridine-oxazoline and related ligands[J]. ACS Catalysis, 2017, 7(6): 4144-4151. |
| [77] | EBI T, SEN A, DHITAL R N, et al. Design of experimental conditions with machine learning for collaborative organic synthesis reactions using transition-metal catalysts[J]. ACS Omega, 2021, 6(41): 27578-27586. |
| [78] | COLEY C W, BARZILAY R, JAAKKOLA T S, et al. Prediction of organic reaction outcomes using machine learning[J]. ACS Central Science, 2017, 3(5): 434-443. |
| [79] | SAINI V, SHARMA A, NIVATIA D. A machine learning approach for predicting the nucleophilicity of organic molecules[J]. Physical Chemistry Chemical Physics, 2022, 24(3): 1821-1829. |
| [80] | PALMER D S, O’BOYLE N M, GLEN R C, et al. Random forest models to predict aqueous solubility[J]. Journal of Chemical Information and Modeling, 2007, 47(1): 150-158. |
| [81] | MAMEDE R, PEREIRA F, AIRES-DE-SOUSA J. Machine learning prediction of UV-Vis spectra features of organic compounds related to photoreactive potential[J]. Scientific Reports, 2021, 11(1): 1-11. |
| [82] | BANERJEE S, SREENITHYA A, SUNOJ R B. Machine learning for predicting product distributions in catalytic regioselective reactions[J]. Physical Chemistry Chemical Physics, 2018, 20(27): 18311-18318. |
| [83] | ZHANG Q Y, AIRES-DE-SOUSA J. Structure-based classification of chemical reactions without assignment of reaction centers[J]. Journal of Chemical Information and Modeling, 2005, 45(6): 1775-1783. |
| [84] | SHIELDS B J, STEVENS J, LI J, et al. Bayesian reaction optimization as a tool for chemical synthesis[J]. Nature, 2021, 590(7844): 89-96. |
| [85] | RYU S, KWON Y, KIM W Y. A Bayesian graph convolutional network for reliable prediction of molecular properties with uncertainty quantification[J]. Chemical Science, 2019, 10(36): 8438-8446. |
| [86] | WANG S W, PILLAI H S, XIN H L. Bayesian learning of chemisorption for bridging the complexity of electronic descriptors[J]. Nature Communications, 2020, 11(1): 1-7. |
| [87] | ZHOU Z P, LI X C, ZARE R N. Optimizing chemical reactions with deep reinforcement learning[J]. ACS Central Science, 2017, 3(12): 1337-1344. |
| [88] | SIMM G N C, PINSLER R, HERNáNDEZ-LOBATO J M. Reinforcement learning for molecular design guided by quantum mechanics[EB/OL]. (2020-02-18)[2022-03-05]. https://arxiv.org/abs/2002.07717. |
| [89] | POPOVA M, ISAYEV O, TROPSHA A. Deep reinforcement learning forde novo drug design[J]. Science Advances, 2018, 4(7): eaap7885. |
| [90] | ATANCE S R, DIEZ J V, ENGKVIST O, et al. De novo drug design using reinforcement learning with graph-based deep generative models[J]. Journal of Chemical Information and Modeling, 2022, 62(20): 4863-4872. |
/
| 〈 |
|
〉 |